‘Single-cell profiling’ is helping neuroscientists see how disease affects major brain cell types and identify common, potentially targetable pathways
After decades of fundamental scientific and drug discovery research, Alzheimer’s disease has remained inscrutable and incurable, with a bare minimum of therapeutic progress. But in a new review article in Nature Neuroscience, MIT scientists write that by employing the new research capability of single-cell profiling, the field has rapidly achieved long-sought insights with strong potential for both explaining Alzheimer’s disease and doing something meaningful about it. By analyzing this new evidence, for instance, the authors show that the disease’s disruptions converge on five main areas of cellular function, or “pathways,” in each of five major brain cell types.
Single-cell profiling technologies produce comprehensive measurements of genetic activity in individual cells, such as levels of RNA which is transcribed from DNA, so that the cell’s functions and roles of in the biology of the brain, and the pathology of disease, can be assessed. Single-cell profiling technologies go beyond genome sequencing, which catalogs the DNA present in most every cell of a person, by revealing how each cell is uniquely making use of that common set of instructions. In studying Alzheimer’s disease, scientists have been using single-cell profiling to see how various brain cells, such as distinct types of neurons and microglia and astrocytes act differently in disease compared to how they behave in a healthy brain.
In the article, MIT Brain and Cognitive Sciences doctoral student Mitch Murdock and Picower Professor Li-Huei Tsai, Director of MIT’s Picower Institute for Learning and Memory and Aging Brain Initiative, write that while the findings of single-cell profiling studies confirm that the disease’s terrible effects are complex and far-reaching, there appear to also be five pathways that become perturbed in each of five major cell types. Investigating these pathways, they write, could produce valuable biomarkers of disease and yield meaningful targets for therapeutic intervention:
- Inflammation and immune response
- Lipid (fat molecule) signaling and metabolism
- Metabolic stress and protein folding
- DNA damage and cellular senescence (aging)
- Interactions with brain vasculature (blood vessels)
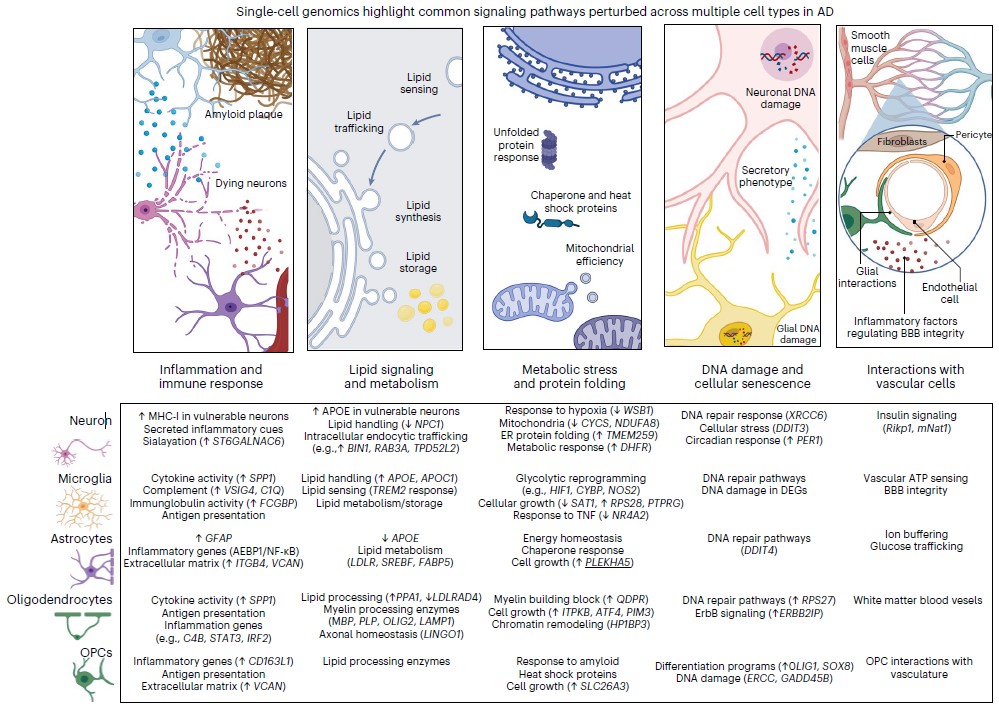
A figure from the paper depicts pathways perturbed in Alzheimer’s disease. Below examples are listed for each pathway by major brain cell type.
For each of these pathways in neurons, microglia, astrocytes, oligodendrocytes and oligodendrocyte precursor cells, Tsai and Murdock identify specific differences in gene regulation, found in single-cell studies, that significantly occur in brains of Alzheimer’s patients or mouse models compared to healthy control samples.
For example, Tsai and Murdock highlight more than a dozen genes all intimately involved in lipid processing whose expression is altered in various ways in various cells in the brain’s prefrontal cortex. For another example they show that all five cell types show impairments in DNA repair, albeit by changed expression of different genes in each.
“By identifying vulnerable cell types and the molecular programs that give rise to them, therapeutic interventions might reverse aberrant cellular trajectories,” Murdock and Tsai wrote in Nature Neuroscience. “While many transcriptional alterations are cell-type specific, these changes ultimately might converge on shared signaling pathways across cell types that might represent targets for new therapeutic strategies.”
To be sure, the authors note, there is still plenty of work to be done, both in refining and improving on single-cell techniques and also exploiting newer related opportunities. The paper notes a number of issues that must be carefully considered in producing valid single-cell profiling results, including where cells are sampled in the brain for sequencing, from whom, and in what condition. Moreover, it’s not always straightforward to show how changes in gene expression necessarily affect biology and it’s even harder to know whether any particular intervention, for instance to target altered inflammation pathways, will prove safe and effective as a therapy.
Future directions, meanwhile could include making greater use of “spatial transcriptomics,” which measures gene transcription in cells where they are situated within the brain, rather than removing them for analysis. Studies should be expanded to incorporate more human samples so that varying disease and demographic differences can be fully accounted for. Datasets should be shared and integrated, the authors write, and better comparisons between human and mouse samples are necessary to better understand how well, or not, they overlap.
“Single-cell profiling facilitates a nuanced portrait of the diverse cellular processes perturbed in the AD brain,” Tsai and Murdock conclude. “These varied molecular programs help explain the divergence between healthy aging and cognitive decline, and highlight cell-type-specific molecular programs involved in AD. Core signaling modules are disrupted across multiple cell types, and manipulating disrupted cellular states will pave the way for new therapeutic opportunities.”