Tactile stimulation improved motor performance, reduced phosphorylated tau, preserved neurons and synapses and reduced DNA damage, a new study shows
Evidence that non-invasive sensory stimulation of 40 Hz gamma frequency brain rhythms can reduce Alzheimer’s disease pathology and symptoms, already shown with light and sound by multiple research groups in mice and humans, now extends to tactile stimulation. A new study by MIT scientists shows that Alzheimer’s model mice exposed to 40 Hz vibration an hour a day for several weeks showed improved brain health and motor function compared to untreated controls.
The MIT group is not the first to show that gamma frequency tactile stimulation can affect brain activity and improve motor function, but they are the first to show that the stimulation can also reduce levels of the hallmark Alzheimer’s protein phosphorylated tau, keep neurons from dying or losing their synapse circuit connections, and reduce neural DNA damage.
“This work demonstrates a third sensory modality that we can use to increase gamma power in the brain,” said Li-Huei Tsai, corresponding author of the study, director of The Picower Institute for Learning and Memory and the Aging Brain Initiative at MIT, and Picower Professor in the Department of Brain and Cognitive Sciences (BCS). “We are very excited to see that 40 Hz tactile stimulation benefits motor abilities, which has not been shown with the other modalities. It would be interesting to see if tactile stimulation can benefit human subjects with impairment in motor function.”
Ho-Jun Suk, Nicole Buie, Guojie Xu and Arit Banerjee are lead authors of the study in Frontiers in Aging Neuroscience and Ed Boyden, Y. Eva Tan Professor of Neurotechnology at MIT, is a co-senior author of the paper. Boyden, an affiliate member of The Picwoer Institute, is also appointed in BCS as well as the Departments of Bioengineering and Media Arts and Sciences, the McGovern Institute for Brain Research, and the K. Lisa Yang Cener for Bionics.
Feeling the vibe
In a series of papers starting in 2016, a collaboration led by Tsai’s lab has demonstrated that light flickering and/or sound clicking at 40 Hz (a technology called GENUS for Gamma Entrainment Using Sensory stimuli), reduces levels of amyloid-beta and tau proteins, prevents neuron death and preserves synapses and even sustains learning and memory in a variety of Alzheimer’s disease mouse models. Most recently in pilot clinical studies the team showed that 40 Hz light and sound stimulation was safe, successfully increased brain activity and connectivity and appeared to produce significant clinical benefits in a small cohort of human volunteers with early-stage Alzheimer’s disease. Other groups have replicated and corroborated health benefits of 40 Hz sensory stimulation and an MIT spin-off company, Cognito Therapeutics, has launched stage III clinical trials of light and sound stimulation as an Alzheimer’s treatment.
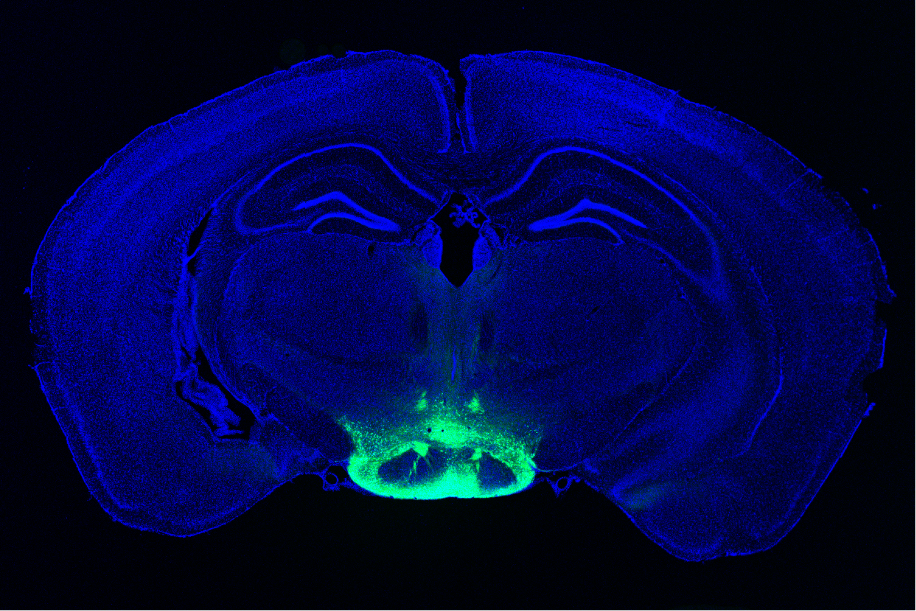
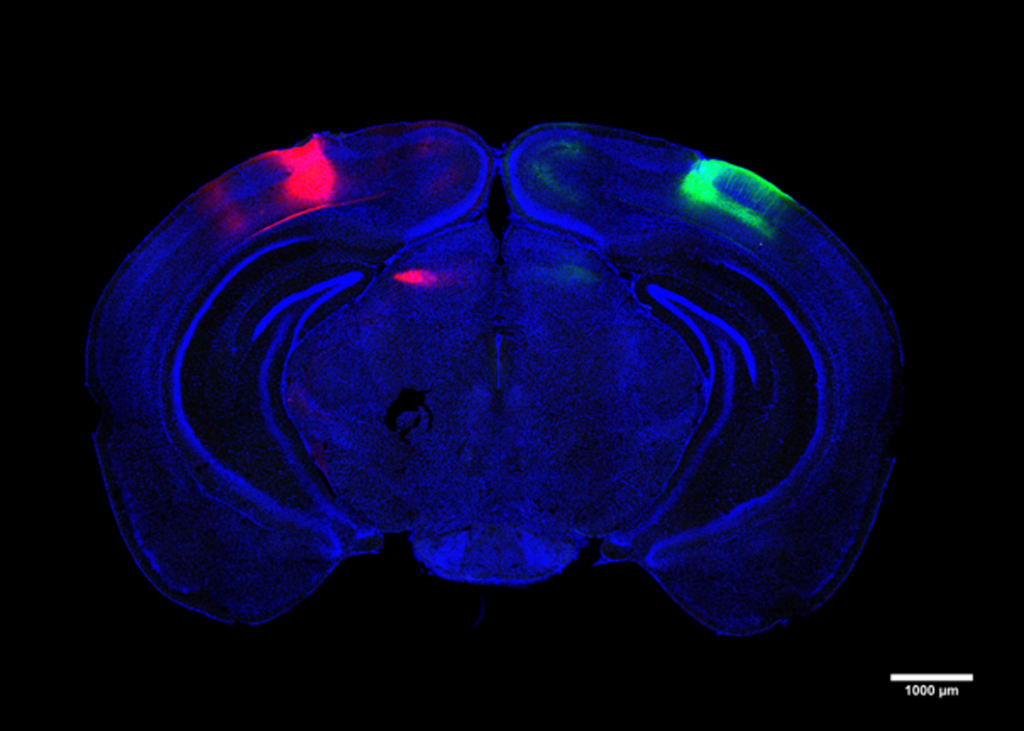
The new study tested whether whole-body 40 Hz tactile stimulation produced meaningful benefits in two commonly used mouse models of Alzheimer’s neurodegeneration, the Tau P301S mouse, which recapitulates the disease’s tau pathology, and the CK-p25 mouse, which recapitulates the synapse loss and DNA damage seen in human disease. The team focused its analyses in two areas of the brain: the primary somatosensory cortex (SSp), where tactile sensations are processed, and the primary motor cortex (MOp), where the brain produces movement commands for the body.
To produce the vibration stimulation, the researchers placed mouse cages over speakers playing 40 Hz sound, which vibrated the cages. Non-stimulated control mice were in cages interspersed in the same room so that all the mice heard the same 40 Hz sound. The differences measured between the stimulated and control mice were therefore made by the addition of tactile stimulation.
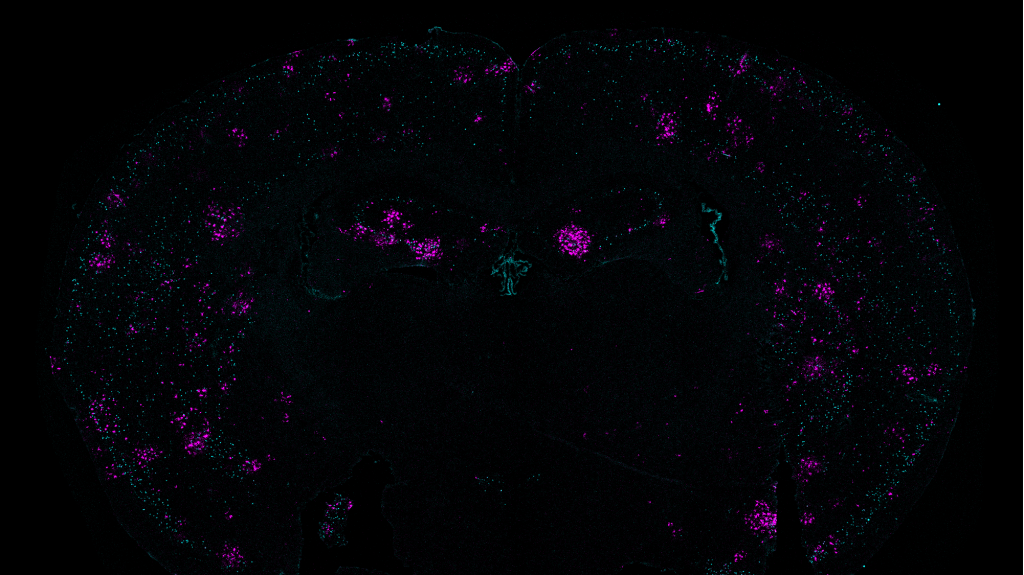
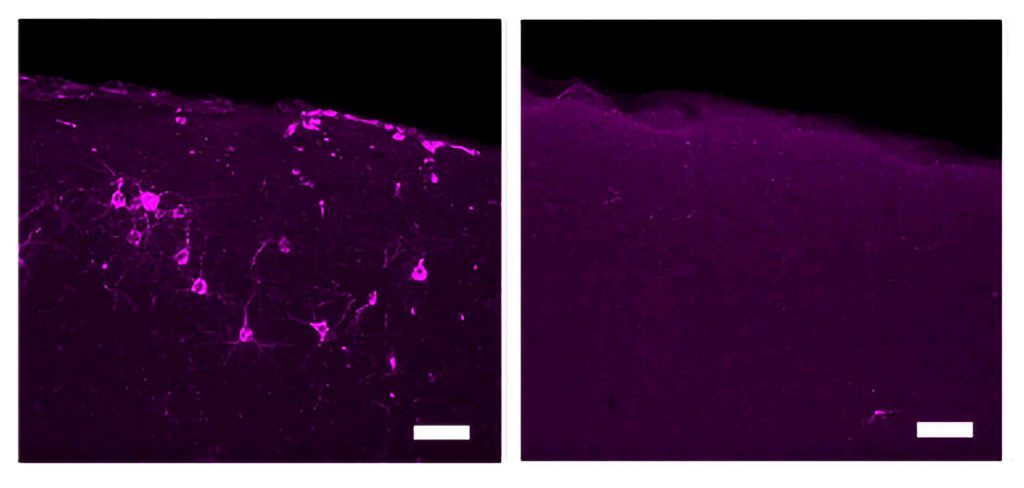
First the researchers confirmed that 40 Hz vibration made a difference in neural activity in the brains of healthy (i.e. non-Alzheimer’s) mice. As measured by expression of c-fos protein, activity increased two-fold in the SSp and more than 3-fold in the MOp, a statistically significant increase in the latter case.
Once the researchers knew that 40 Hz tactile stimulation could increase neural activity, they assessed the impact on disease in the two mouse models. To ensure both sexes were represented, the team used male P301S mice and female CK-p25 mice.
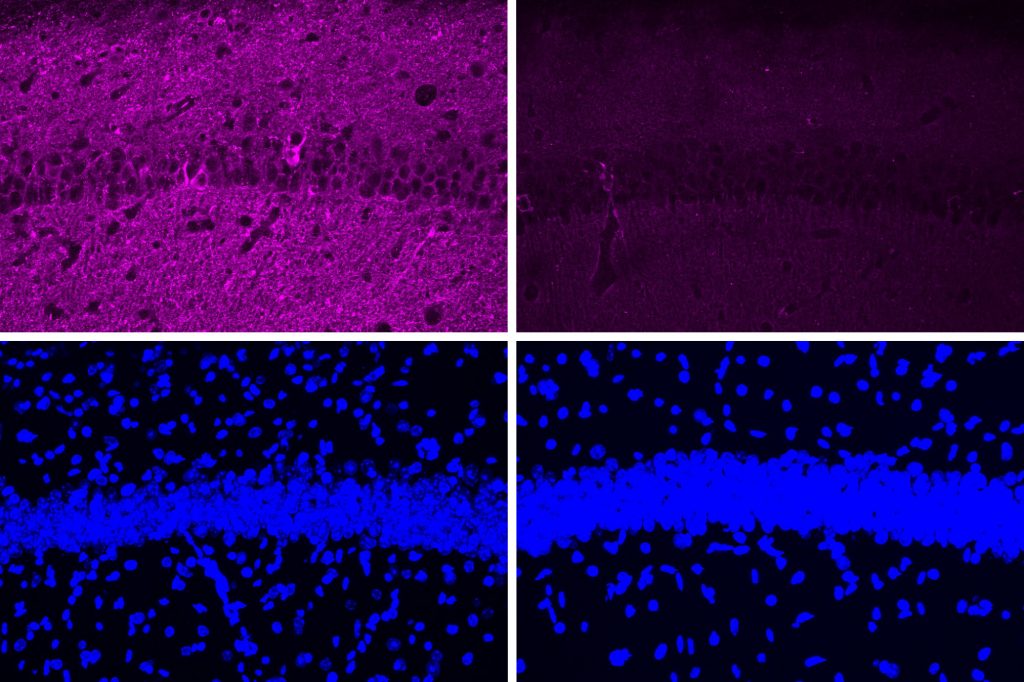
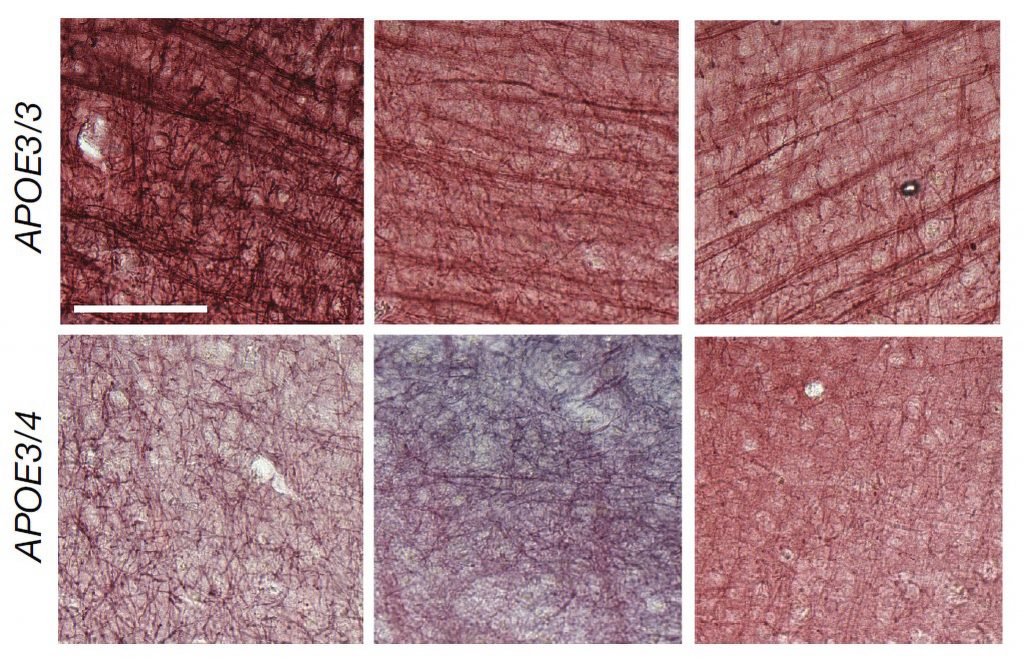
P301S mice stimulated for three weeks showed significant preservation of neurons compared to unstimulated controls in both brain regions. Stimulated mice also showed significant reductions in tau in the SSp by two measures, and exhibited similar trends in the MOp.
CK-p25 mice received six weeks of vibration stimulation. These mice showed higher levels of synaptic protein markers in both brain regions compared to unvibrated control mice. They also showed reduced levels of DNA damage.
Finally the team assessed the motor abilities of mice exposed to the vibration vs. not exposed. They found that both mouse models were able to stay on a rotating rod significantly longer. P301S mice also hung on to a wire mesh for significantly longer than control mice while CK-p25 mice showed a positive, though non-significant trend.
“The current study, along with our previous studies using visual or auditory GENUS demonstrates the possibility of using non-invasive sensory stimulation as a novel therapeutic strategy for ameliorating pathology and improving behavioral performance in neurodegenerative diseases,” the authors concluded.
Support for the study came from The JPB Foundation, The Picower Institute for Learning and Memory, Eduardo Eurnekian, The DeGroof-VM Foundation, Halis Family Foundation, Melissa and Doug Ko Hahn, Lester Gimpelson, Eleanor Schwartz Charitable Foundation, The Dolby Family, Kathleen and Miguel Octavio, Jay and Carroll Miller, Anne Gao and Alex Hu and Charles Hieken.