One of the most significant genetic risk factors for developing Alzheimer’s disease is a gene called APOE4, which is carried by almost half of all Alzheimer’s patients. A new study from MIT shows that this gene has widespread effects on brain cells’ ability to metabolize lipids and respond to stress.
In studies of human brain cells and yeast cells, the researchers found that the APOE4 gene significantly disrupts brain cells’ ability to carry out their normal functions. They also showed that treating these cells with extra choline, a widely available supplement that is considered safe for human use, could reverse many of these effects.
The researchers hope that their findings will lead to clinical studies of choline in people who carry the APOE4 gene, who make up about 14 percent of the overall population. Previous trials looking at choline’s effects on cognition showed mixed results, but those trials were not targeted specifically to people with the APOE4 gene.
“What we would really like to see is whether in the human population, in those APOE4 carriers, if they take choline supplements to a sufficient amount, whether that would delay or give them some protection against developing dementia or Alzheimer’s disease,” says Li-Huei Tsai, the director of MIT’s Picower Institute for Learning and Memory.
Tsai and the late Susan Lindquist, former director of MIT’s Whitehead Institute for Biomedical Research, are the senior authors of the study, which appears today in Science Translational Medicine. The paper’s three lead authors are former Whitehead and MIT postdocs Grzegorz Sienski and Priyanka Narayan, and current MIT postdoc Julia Maeve Bonner.
Lipid dysregulation
The human gene for APOE, or apolipoprotein E, comes in three versions. While APOE4 is linked to higher risk for Alzheimer’s, APOE2 is considered protective, and APOE3, the most common variant, is neutral.
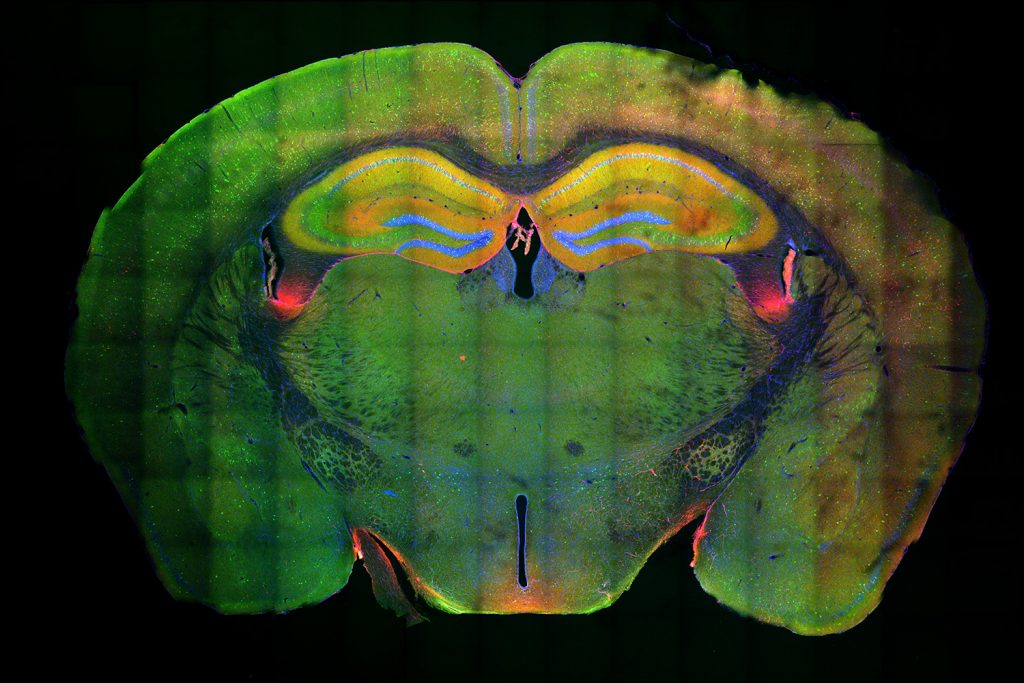
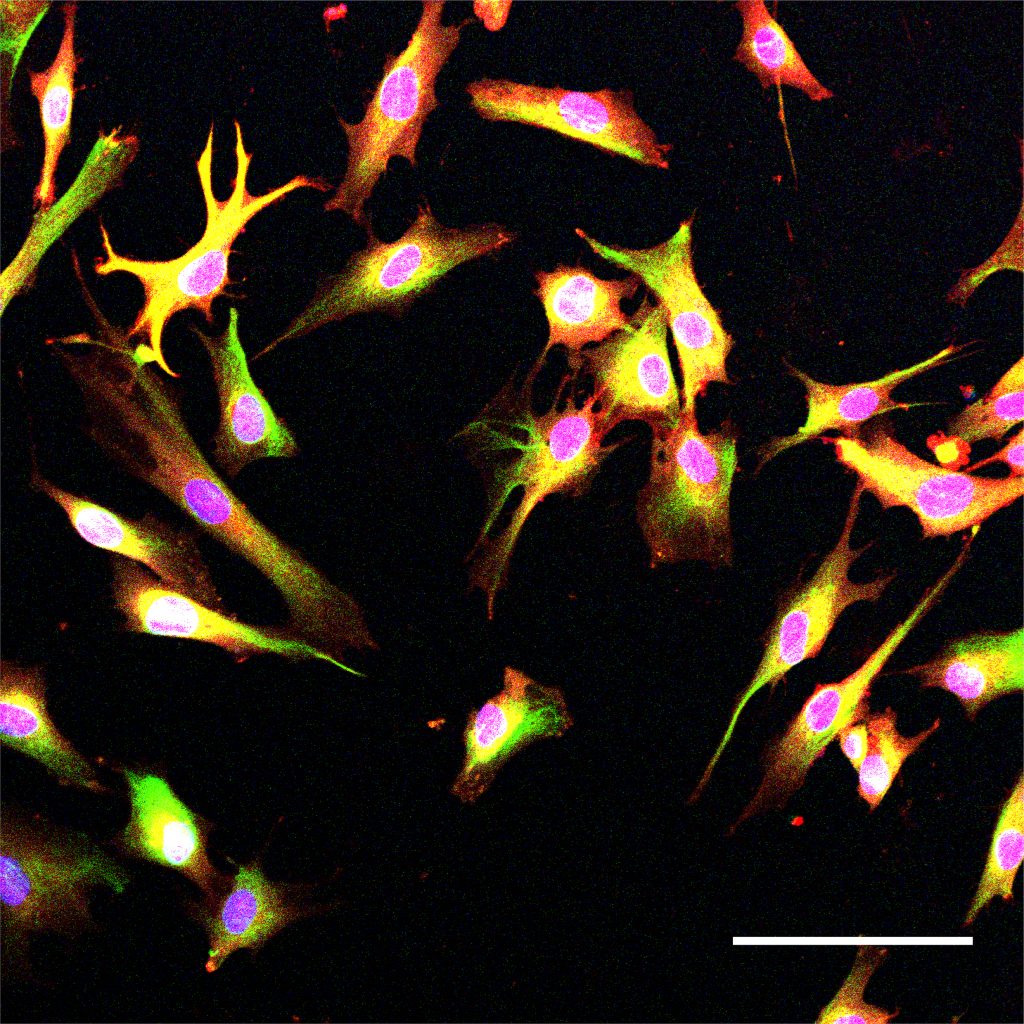
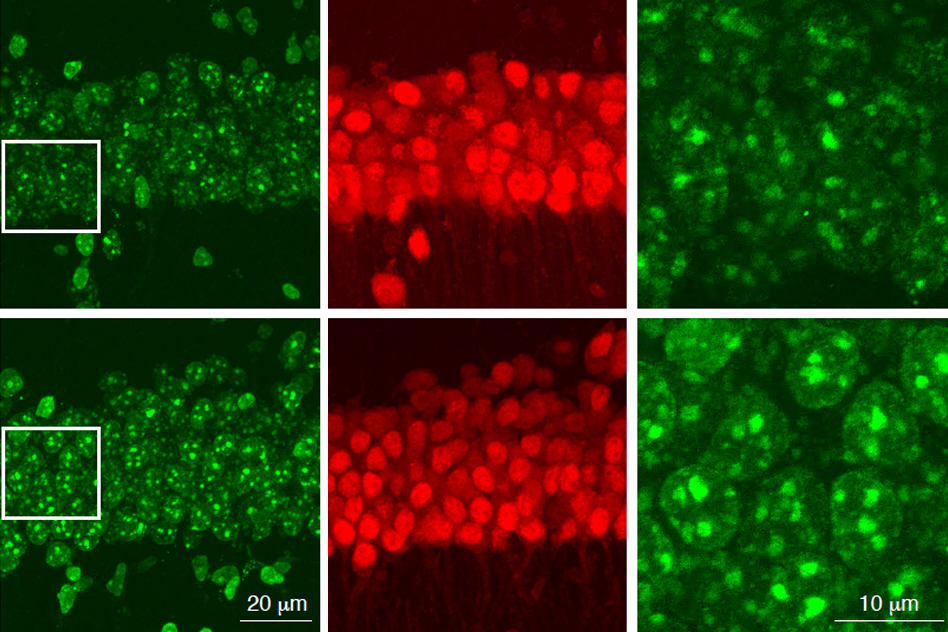
APOE is known to be involved in lipid metabolism, but its role in the development of Alzheimer’s has been unclear, Tsai says. To try to learn more about this connection, the researchers created human induced pluripotent stem cells that carry either the APOE3 or APOE4 gene in an otherwise identical genetic background. They then stimulated these cells to differentiate into astrocytes, the brain cells that produce the most APOE.
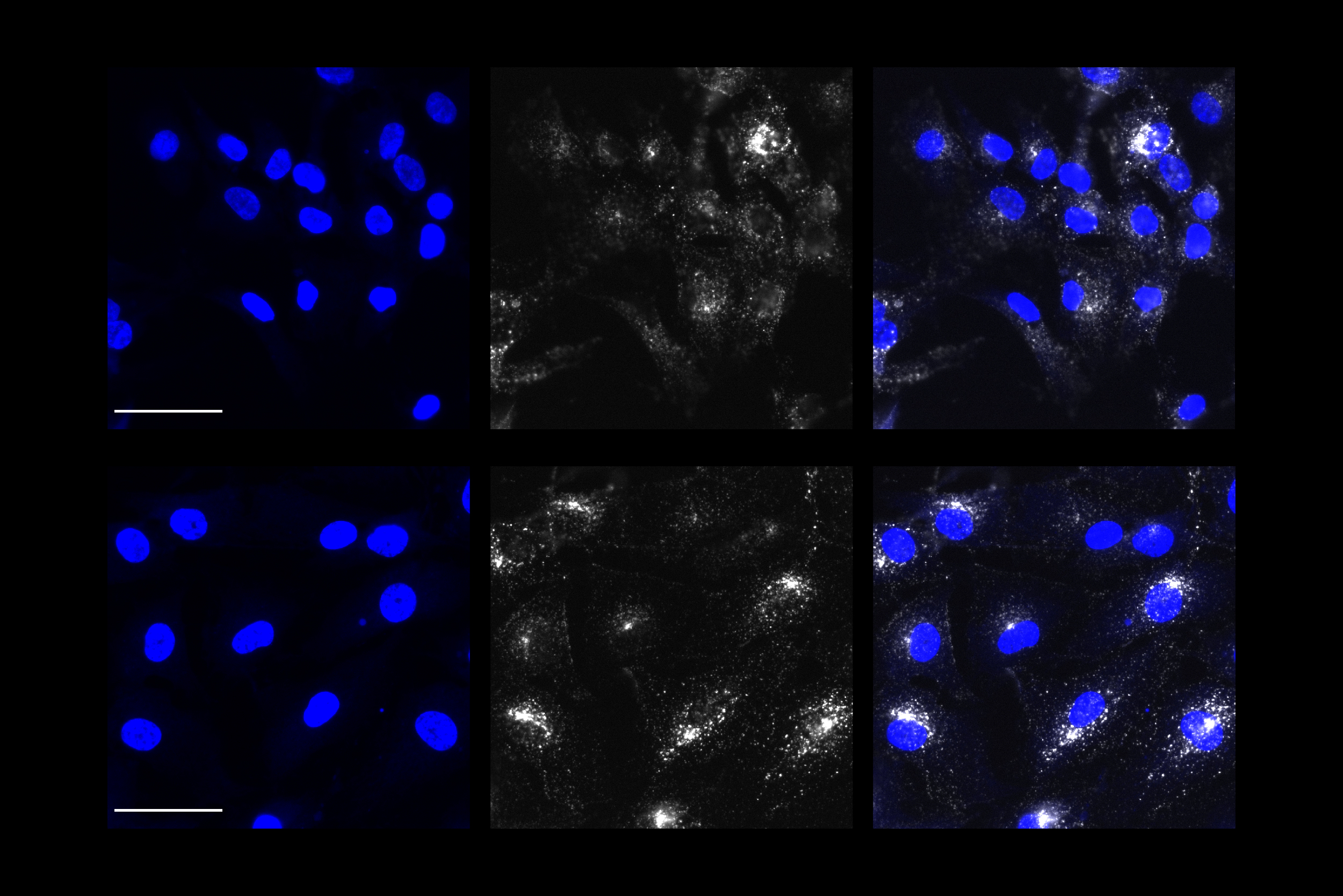
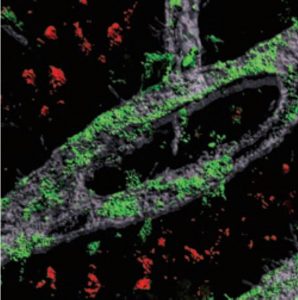
APOE4 astrocytes showed dramatic changes in how they process lipids compared to APOE3. In APOE4 astrocytes, there was a significant buildup of neutral lipids and cholesterol. These astrocytes also accumulated droplets containing a type of lipids called triglycerides, and these triglycerides had many more unsaturated fatty acid chains than normal. These changes all disrupt the normal lipid balance inside the cells. The authors also noted APOE4-dependent lipid disruptions in another important brain cell, microglia.
“When lipid homeostasis is compromised, then a lot of very essential processes are affected, such as intracellular trafficking, vesicular trafficking, and endocytosis. A lot of the cells’ essential functions are compromised,” Tsai says.
“This balance is really important for cells to be able to perform normal functions like generate membranes and so on, but also to be able to absorb stress,” Bonner says. “We think that one of the things that’s happening is that these cells are less able to absorb stress because they’re already in this heightened lipid dysregulation state.”
The researchers also found that yeast cells engineered to express the human version of APOE4 showed many of the same defects. Using these cells, they performed a systematic genetic screen to determine the molecular basis of the defects seen in APOE4 cells. This screen showed that turning on a pathway that normally produces phospholipids, an essential component of cell membranes, can reverse some of the damage seen in APOE4 cells. This suggests that APOE4 somehow increases the requirement for phospholipid synthesis.
The researchers also found that growing APOE4 yeast cells on a very nutrient-rich growth medium helped them to survive better than APOE4 yeast cells grown on the typical growth medium. Further experiments revealed that the nutrient that helped APOE4 cells survive is choline, a building block that cells use to make phospholipids. The researchers then treated their human APOE4 astrocyte cells with choline to promote phospholipid synthesis, and found that it also reversed much of the damage they had seen in those cells, including the accumulation of cholesterol and lipid droplets.
Choline deficiency
The researchers have now begun studying a mouse model of Alzheimer’s that is also engineered to express the human APOE4 gene. They hope to investigate whether choline can help to reverse some of the symptoms of Alzheimer’s in these mice.
Choline is naturally found in foods such as eggs, meat, fish, and some beans and nuts. The minimum recommended intake of choline is 550 milligrams per day for men and 425 milligrams per day for women, but most people don’t consume that much, Tsai says. The new study offers preliminary evidence that people who carry the APOE4 gene may benefit from taking choline supplements, she says, although clinical trials are necessary to confirm that.
“What our results suggest is that if you are an APOE2 or APOE3 carrier, even you are somewhat choline deficient you can cope with it,” Tsai says. “But if you are an APOE4 carrier, then if you don’t take enough choline, then that will have a more dire consequences. The APOE4 carriers are more susceptible to choline deficiency.”
The research was funded by the EMBO Fellowship, the Helen Hay Whitney Foundation, the National Institutes of Health, the Damon Runyon Foundation, the Neurodegeneration Consortium, the Robert A. and Renee E. Belfer Foundation, the Howard Hughes Medical Institute, the Ludwig Family Foundation, and Kara and Stephen Ross.
–From MIT News