Stimulating gamma brain waves may protect cancer patients from memory impairment and other cognitive effects of chemotherapy.
Patients undergoing chemotherapy often experience cognitive effects such as memory impairment and difficulty concentrating — a condition commonly known as “chemo brain.”
MIT researchers have now shown that a noninvasive treatment that stimulates gamma frequency brain waves may hold promise for treating chemo brain. In a study of mice, they found that daily exposure to light and sound with a frequency of 40 hertz protected brain cells from chemotherapy-induced damage. The treatment also helped to prevent memory loss and impairment of other cognitive functions.
This treatment, which was originally developed as a way to treat Alzheimer’s disease, appears to have widespread effects that could help with a variety of neurological disorders, the researchers say.
“The treatment can reduce DNA damage, reduce inflammation, and increase the number of oligodendrocytes, which are the cells that produce myelin surrounding the axons,” says Li-Huei Tsai, director of MIT’s Picower Institute for Learning and Memory and Picower Professor in the MIT Department of Brain and Cognitive Sciences. “We also found that this treatment improved learning and memory, and enhanced executive function in the animals.”
Tsai, who also leads MIT’s Aging Brain Initiative, is the senior author of the new study, which appears today in Science Translational Medicine. The paper’s lead author is TaeHyun Kim, an MIT postdoc.
Protective brain waves
Several years ago, Tsai and her colleagues began exploring the use of light flickering at 40 hertz (cycles per second) as a way to improve the cognitive symptoms of Alzheimer’s disease. Previous work had suggested that Alzheimer’s patients have impaired gamma oscillations — brain waves that range from 25 to 80 hertz (cycles per second) and are believed to contribute to brain functions such as attention, perception, and memory.
Tsai’s studies in mice have found that exposure to light flickering at 40 hertz or sounds with a pitch of 40 hertz can stimulate gamma waves in the brain, which has many protective effects, including preventing the formation of amyloid beta plaques. Using light and sound together provides even more significant protection. The treatment also appears promising in humans: Phase 1 clinical trials in people with early-stage Alzheimer’s disease have found the treatment is safe and does offer some neurological and behavioral benefits.
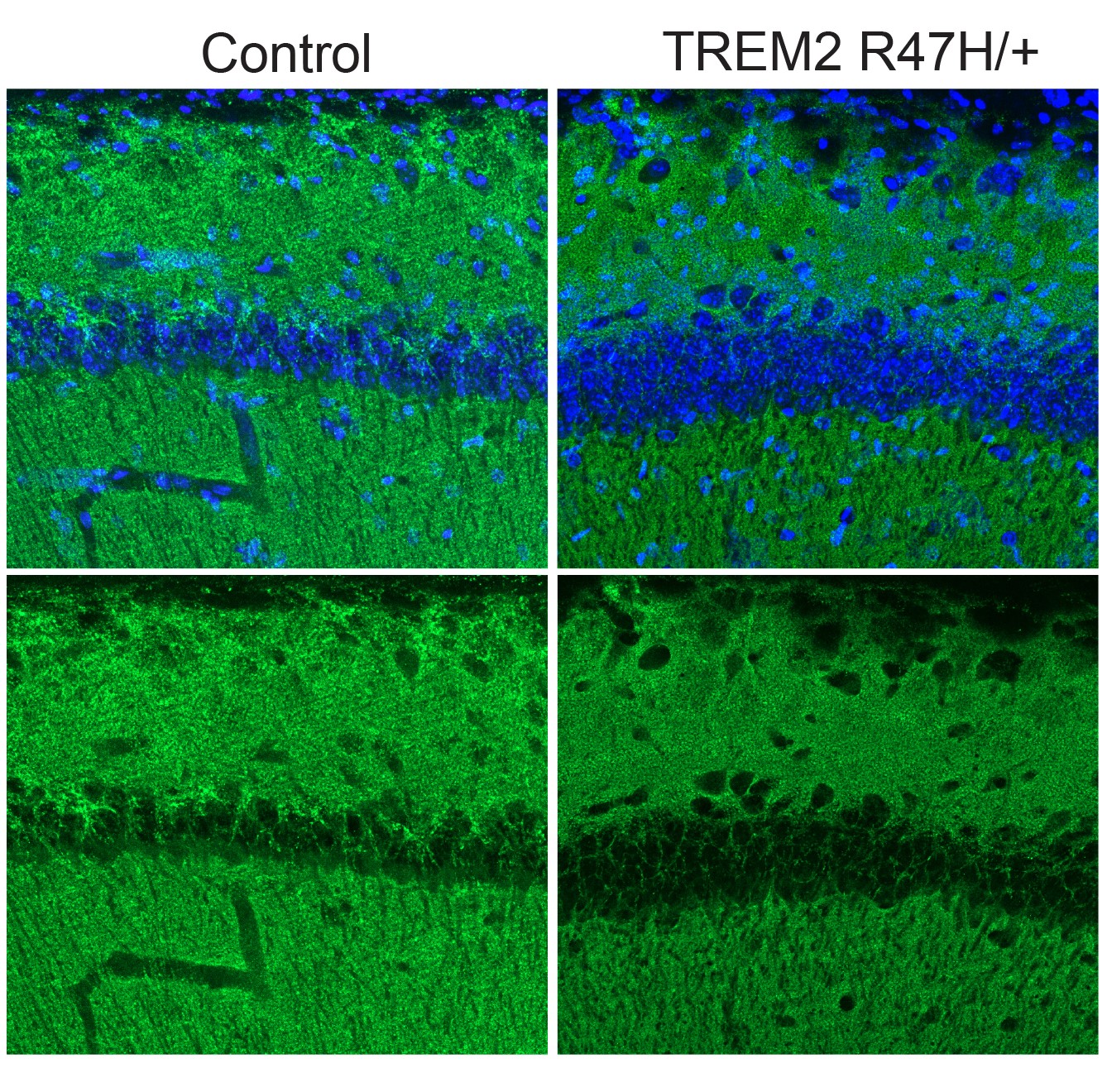
In the new study, the researchers set out to see whether this treatment could also counteract the cognitive effects of chemotherapy treatment. Research has shown that these drugs can induce inflammation in the brain, as well as other detrimental effects such as loss of white matter — the networks of nerve fibers that help different parts of the brain communicate with each other. Chemotherapy drugs also promote loss of myelin, the protective fatty coating that allows neurons to propagate electrical signals. Many of these effects are also seen in the brains of people with Alzheimer’s.
“Chemo brain caught our attention because it is extremely common, and there is quite a lot of research on what the brain is like following chemotherapy treatment,” Tsai says. “From our previous work, we know that this gamma sensory stimulation has anti-inflammatory effects, so we decided to use the chemo brain model to test whether sensory gamma stimulation can be beneficial.”
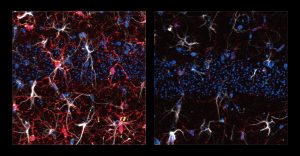
As an experimental model, the researchers used mice that were given cisplatin, a chemotherapy drug often used to treat testicular, ovarian, and other cancers. The mice were given cisplatin for five days, then taken off of it for five days, then on again for five days. One group received chemotherapy only, while another group was also given 40-hertz light and sound therapy every day.
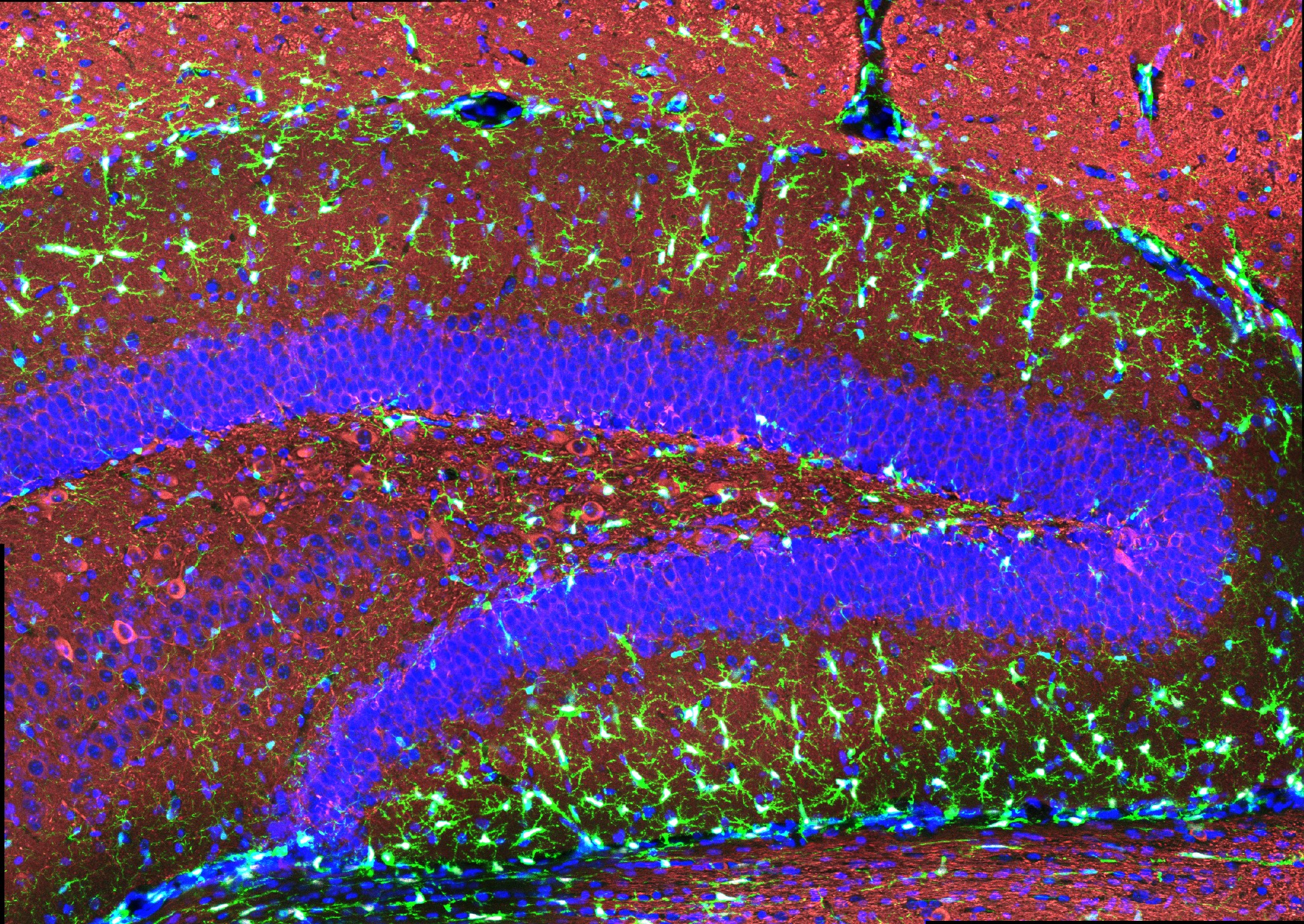
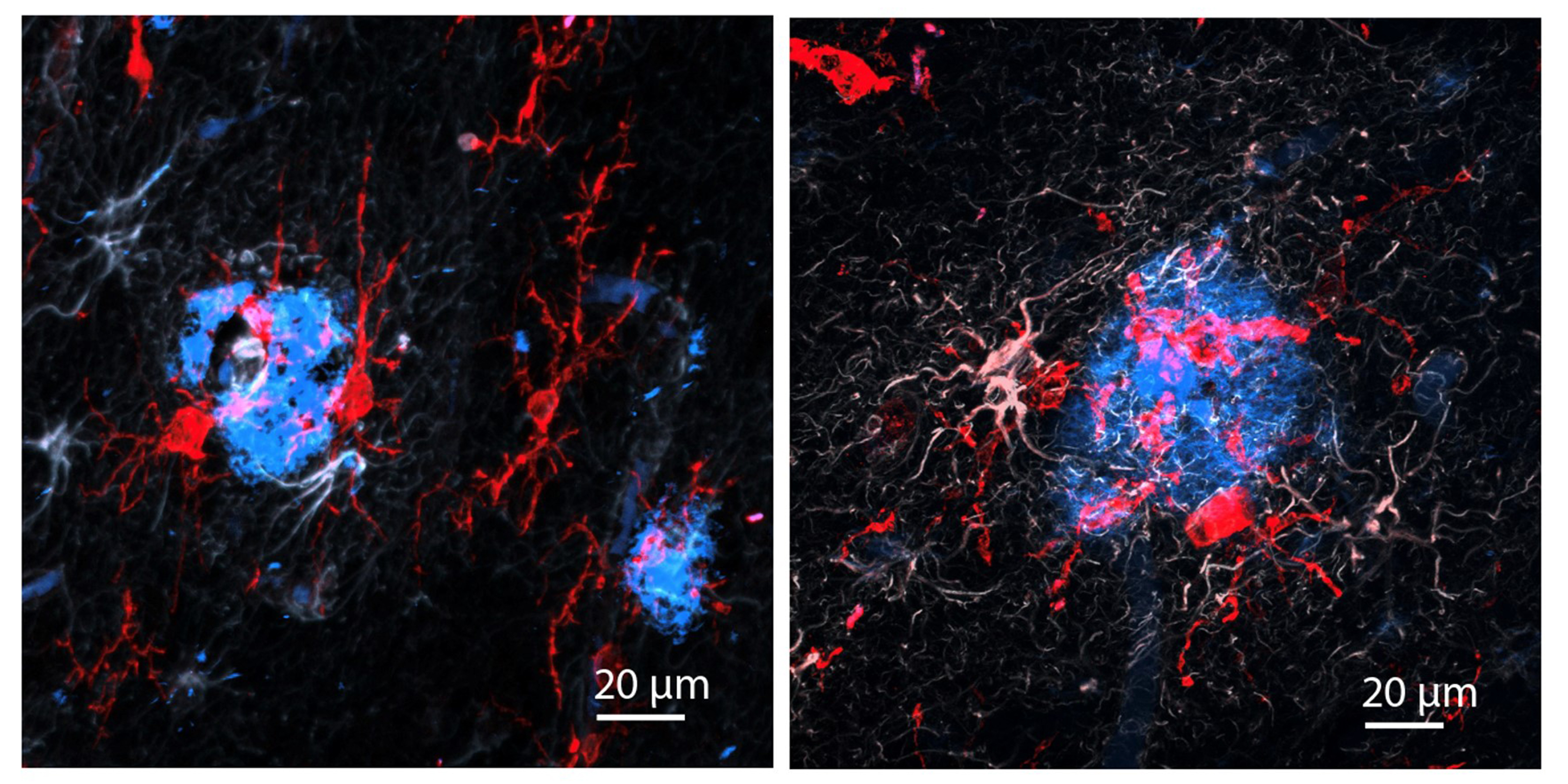
After three weeks, mice that received cisplatin but not gamma therapy showed many of the expected effects of chemotherapy: brain volume shrinkage, DNA damage, demyelination, and inflammation. These mice also had reduced populations of oligodendrocytes, the brain cells responsible for producing myelin.
However, mice that received gamma therapy along with cisplatin treatment showed significant reductions in all of those symptoms. The gamma therapy also had beneficial effects on behavior: Mice that received the therapy performed much better on tests designed to measure memory and executive function.
“A fundamental mechanism”
Using single-cell RNA sequencing, the researchers analyzed the gene expression changes that occurred in mice that received the gamma treatment. They found that in those mice, inflammation-linked genes and genes that trigger cell death were suppressed, especially in oligodendrocytes, the cells responsible for producing myelin.
In mice that received gamma treatment along with cisplatin, some of the beneficial effects could still be seen up to four months later. However, the gamma treatment was much less effective if it was started three months after the chemotherapy ended.
The researchers also showed that the gamma treatment improved the signs of chemo brain in mice that received a different chemotherapy drug, methotrexate, which is used to treat breast, lung, and other types of cancer.
“I think this is a very fundamental mechanism to improve myelination and to promote the integrity of oligodendrocytes. It seems that it’s not specific to the agent that induces demyelination, be it chemotherapy or another source of demyelination,” Tsai says.
Because of its widespread effects, Tsai’s lab is also testing gamma treatment in mouse models of other neurological diseases, including Parkinson’s disease and multiple sclerosis. Cognito Therapeutics, a company founded by Tsai and MIT Professor Edward Boyden, has finished a phase 2 trial of gamma therapy in Alzheimer’s patients, and plans to begin a phase 3 trial this year.
“My lab’s major focus now, in terms of clinical application, is Alzheimer’s; but hopefully we can test this approach for a few other indications, too,” Tsai says.
The research was funded by the JPB Foundation, the Ko Hahn Seed Fund, and the National Institutes of Health.
–From MIT News